



Разработка новых химических препаратов, с целью облегчения состояния больных при нейродегенеративных заболеваниях, является важным и актуальным направлением современной медицинской химии. Прежде всего, это относится к болезни Альцгеймера (БА) – наиболее часто встречающемуся типу деменции у пожилых людей. По состоянию на 2020 год во всем мире насчитывалось около 50 миллионов человек с БА [1]. Этой болезнью страдают около 6% людей в возрасте 65 лет и старше [2].
Множество молекулярных объектов задействованы в развитии болезни Альцгеймера. Это определяет большой выбор мишеней для QSAR-поиска лекарств, облегчающих течение заболевания. В настоящее время используются: донепезил, галантамин, ривастигмин (ингибиторы ацетилхолинэстеразы) и мемантин (неконкурентный блокатор NMDA-подтипа глутаматных рецепторов) [3]. Поиск новых перспективных соединений связан с разработкой препаратов, способных действовать одновременно на разные основные мишени, вовлеченные в патогенез заболевания.
Для этого требуются усилия многих научных коллективов, врачей клинической практики, огромные финансовые и временные затраты. Снижение издержек возможно при использовании модельных экспериментов или расчетных методов оценки и отбора перспективных соединений. Примеры положительной практики расчетного метода QSAR описаны в литературе [4].
Наша предыдущая работа [5] была посвящена созданию компьютерных моделей активности конъюгатов γ-карболинов и фенотиазина по отношению к ингибированию бутирилхолинэстеразы. Выбор соединений был обусловлен тем, что они одновременно являются селективными ингибиторами BChE и блокаторами NMDA рецепторов [6]. С учетом того, что некоторые производные карбазола (например, аминотетрагидрокарбазолы) способны к модификации течения БА [7], перспективным направлением выглядит модификация конъюгатов путем замены фенотиазиновых фрагментов на карбазольные фрагменты.
Целью настоящей работы явилось создание компьютерных моделей активности конъюгатов γ-карболинов и карбазолов по отношению к ингибированию бутирилхолинэстеразы.
Материалы и методы исследования
Выборка соединений для обучения содержала 15 веществ (рис. 1, табл.). Она была сформирована на основе работы [8]. В таблице представлены данные по ингибированию бутирилхолинэстеразы (EC 3.1.1.8 из лошадиной сыворотки). В качестве меры ингибирующей активности использовали log(1/IC50), где IC50 (мкМ) – концентрация вещества, вызывающая 50% ингибирование BChE.
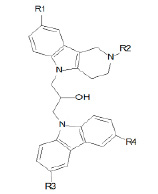
Рис. 1. Конъюгаты γ-карболинов и производных карбазола
Ингибиторная активность конъюгатов γ-карболинов с производными карбазола по отношению к BChE
|
Номер |
R1 |
R2 |
R3 |
R4 |
IC50, мкМ |
log(1/IC50) |
|
1 |
H |
CH3 |
H |
H |
3.40 ± 0.04 |
-0,53 |
|
2 |
H |
C2H5 |
H |
H |
2.19 ± 0.05 |
-0,34 |
|
3 |
CH3 |
CH3 |
H |
H |
3.44 ± 0.34 |
-0,54 |
|
4 |
CH3 |
C2H5 |
H |
H |
6.11 ± 0.08 |
-0,79 |
|
5 |
F |
CH3 |
H |
H |
3.14 ± 0.02 |
-0,50 |
|
6 |
F |
C2H5 |
H |
H |
4.05 ± 0.03 |
-0,61 |
|
7 |
H |
CH3 |
CI |
CI |
3.48 ± 0.69 |
-0,54 |
|
8 |
CH3 |
CH3 |
CI |
CI |
16.4 ± 1.6 |
-1,21 |
|
9 |
CH3 |
C2H5 |
CI |
CI |
12.5 ± 0.8 |
-1,1 |
|
10 |
F |
CH3 |
CI |
CI |
1.93 ± 0.03 |
-0,29 |
|
11 |
F |
C2H5 |
CI |
CI |
33.4 ± 2.2 |
-1,52 |
|
12 |
H |
CH3 |
Br |
Br |
2.69 ± 0.25 |
-0,43 |
|
13 |
H |
C2H5 |
Br |
Br |
21.6 ± 2.10 |
-1,33 |
|
14 |
CH3 |
C2H5 |
Br |
Br |
3.16 ± 0.21 |
-0,50 |
|
15 |
F |
CH3 |
Br |
Br |
1.18 ± 0.06 |
-0,07 |
Структуры молекул описывали двумерными (2D) и трехмерными (3D) QSAR-дескрипторами. В качестве 2D-дескрипторов использовали 45 физико-химических характеристик, рассчитанных на базе компьютерного комплекса программ HYBOT [9]. Для расчета 3D-дескрипторов был проведен полный конформационный анализ с использованием программы Cache Worksystem Pro 6.0. Методика расчета дескрипторов на базе спектров межатомных внутримолекулярных взаимодействий (СМВВ) [10; 11] была описана в работе [5]. Для дальнейшего анализа использовали парные атом – атомные взаимодействия с участием: 1) Н-донор – Н-донор (DDF, DDE); 2) Н-акцептор – Н-акцептор (AAF, AAE); 3) Н-донор – Н-акцептор (DAF, DAE); 4) положительно заряженных атомов; 5) отрицательно заряженных атомов; 6) положительно и отрицательно заряженных атомов, а также стерические (Ван-дер-Ваальсовое) взаимодействия атомов (VDW).
Таким образом, для описания пространственной структуры каждого соединения с помощью СМВВ использовали 10*100=1000 дескрипторов СМВВ. Общее количество дескрипторов, использованных в работе, составляло 1045.
Отбор дескрипторов осуществлялся через анализ корреляционной матрицы. Для этого использовали итерационную процедуру [5]. В результате число дескрипторов сократилось с 1045 до 208. Для нахождения связи между структурой веществ и их ингибиторной активностью был привлечён метод множественной линейной регрессии, реализованный в программе SVD [12]. Применяли только внутреннее тестирование, на основе кросс-валидации с выбором по пять (10 итераций). Это обусловлено малой величиной обучающей выборки. Использовали статистические характеристики QSAR-моделей: n – число соединений; R2 – коэффициент линейной корреляции; s – стандартное отклонение; FIT – модифицированный критерий Фишера [13]; R2p – рандомизационный параметр [14]. Формирование моделей проводили с помощью полного перебора комбинаций из 1-3 дескрипторов. Лучшие модели были отобраны на основе статистики FIT. Для оценки области применимости (AD) использовали интервальный метод.
Результаты исследования и их обсуждение
Две лучшие регрессионные модели, представленные ниже:
log(1/IC50) = 1.78(±0.54) – 22.0(±5.6) AAF-40(7.8-8.0) –
– 4.74(±1.00) DAF-28(5.4-5.6) – 0.424(±0.102) VDW-57(11.2-11.4) (1)
n=15; R2=0.794; s=0.21; FIT=1.77; R2cv=0.654; scv=0.28; FITcv=0.87; R2p=0.674
AD: AAF-40(7.8-8.0) = 0.0005÷0.0606; DAF-28(5.4-5.6) = 0.007÷0.217; VDW-57(11.2-11.4) = 1.947÷5.138
где AAF-40(7.8-8.0) – интеграл спектра акцептор – акцепторных внутримолекулярных взаимодействий по типу водородной связи на интервале 7.8-8.0 ангстрема; DAF-28(5.4-5.6) – интеграл донор – акцепторных взаимодействий по типу водородной связи на интервале 5.4-5.6 ангстрем; VDW-57(11.2-11.4) – интеграл Ван-дер-Ваальсовых взаимодействий на интервале 11.2-11.4 ангстрема.
log(1/IC50) = 7.81(±1.25) – 0.263(±0.047) VDW-27(5.2-5.4) –
– 0.263(±0.045) VDW-37(7.2-7.4) + 0.219(±0.078) VDW-66(13.0-13.2) (2)
n=15; R2=0.812; s=0.20; FIT=1.99; R2cv=0.649; scv=0.28; FITcv=0.85; R2p=0.696
AD: VDW-27(5.2-5.4) =17.54÷21.93; VDW-37(7.2-7.4) = 11.15÷15.88; VDW-66(13.0-13.2) = 0.03÷3.01
где VDW-27(5.2-5.4) , VDW-37(7.2-7.4) , VDW-66(13.0-13.2) – интегралы спектра Ван-дер-Ваальсовых внутримолекулярных взаимодействий на интервалах 5.2-5.4; 7.2-7.4 и 13.0-13.2 ангстрема соответственно.
Отметим, что обе QSAR-модели имеют удовлетворительные статистические характеристики. Также нужно подчеркнуть, что в лучшие финальные модели (1) и (2) вошли только 3D-дескрипторы, т.е. они являются в нашем случае более информативными и адекватными по сравнению с использованными 2D-дескрипторами. Путем расчета MLR-моделей на основе нормированных дескрипторов были оценены относительные вклады коэффициентов. Согласно модели (1) основной вклад в ингибирование BChE дают атомы, находящиеся на расстоянии 7.8-8.0 ангстрема и проявляющие акцептор – акцепторные взаимодействия (вклад 36.5%), а также атомы, находящиеся на расстоянии 5.4-5.6 ангстрема и проявляющие донор – акцепторные взаимодействия по типу водородной связи (вклад 38.5%). Менее значимый вклад (25.0%) вносят Ван-дер-Ваальсовы взаимодействия атомов на расстоянии 11.2-11.4 Å. При этом следует отметить, что рассмотренные выше дескрипторы характеризуются только отрицательным вкладом в активность. Согласно модели (2) активность может быть описана только с помощью Ван-дер-Ваальсовых взаимодействий атомов, находящихся на расстояниях 5.2-5.4; 7.2-7.4 и 13.0-13.2 Å. Важно подчеркнуть, что в модели (2) дескрипторы вносят разнонаправленный вклад в активность: VDW-27 (39.8%) и VDW-37 (40.7%) понижают активность, а VDW-66 (19.5%) повышает активность.
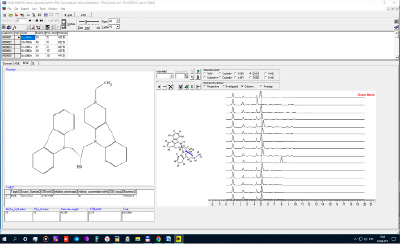
Рис. 2. DAF (донорно-акцепторные) спектры (СМВВ) конъюгатов γ-карболинов и карбазолов
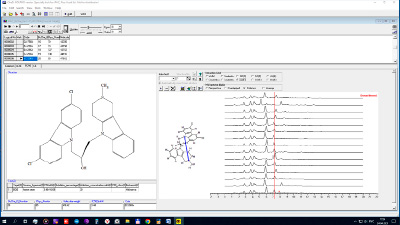
Рис. 3. AAF (акцептор – акцепторные) спектры (СМВВ) конъюгатов γ-карболинов и карбазолов
Рисунки 2 и 3 иллюстрируют вклады в изучаемую активность тех атомов конъюгатов, которые формируют донор – акцепторные и акцептор – акцепторные внутримолекулярные взаимодействия (модель (1)). На этих рисунках показаны результаты работы программы 3D-MOLTRA: слева – 2D-структура конъюгата, в центре его 3D-структура, а справа представлены 15 DAF спектров всех молекул выборки, расположенные колонкой (рис. 2), или 15 AAF спектров (рис. 3). Основной вклад в дескриптор DAF-28(5.4-5.6) СМВВ вносят донор – акцепторные взаимодействия атомов Н гидроксильной группы и азота шестичленного насыщенного кольца γ-карболина, а основной вклад в AAF-40(7.8-8.0) СМВВ вносят акцептор – акцепторные взаимодействия азота карбазола и азота насыщенного цикла γ-карболина.
Выводы
Для конъюгатов γ-карболинов и карбазолов заряд-зарядовые СМВВ не существенны, в отличие от конъюгатов γ-карболинов и фенотиазинов.
Ван-дер-Ваальсовые взаимодействия СМВВ важны в обоих типах конъюгатов. Однако диапазон расстояния, на котором расположены взаимодействующие атомы, несколько отличен (конъюгаты γ-карболинов и фенотиазинов: 5.8-6.0 Å, конъюгаты γ-карболинов и карбазолов: 5.2-5.4; 7.2-7.4 и 13.0-13.2 Å).
Важные акцептор – акцепторные взаимодействия СМВВ несколько отличаются для разных типов конъюгатов. Если для конъюгатов γ-карболинов и карбазолов важны взаимодействия атома азота карбазола и атома азота насыщенного цикла γ-карболина (7.8-8.0 Å), то для конъюгатов γ-карболинов и фенотиазинов на первый план выходят AAF между атомом азота шестичленного насыщенного цикла γ-карболина и атомом кислорода карбонильной группы (7.6-7.8 Å), а также AAF между атомом серы фенотиазина и атомом кислорода карбонильной группы (5.0-5.2 Å).